Fast and accurate deduction of peptides (and proteins) present in a biological mixture such as a tumor, saliva or fungus sample has been the cornerstone in computational omics research for over 30 years. The biological mixture is exposed to a shotgun proteomics experiment where the (long) protein chains in the mixture are decomposed to (smaller) peptides through proteolysis followed by liquid chromatography coupled 2-stage mass-spectrometry where the peptides are separated, and ionized, fragmented and detected. The result of this experiment is that for each peptide present in the mixture, a mass-spectrometry fingerprint (called a MS/MS spectrum or simply ‘spectrum’) is generated. Millions of raw, noisy spectra are produced, in a span of few hours, using modern mass spectrometry (MS) technologies producing several gigabytes of experimental data (See Figure 1). The most commonly employed computational technique to identify peptides from the experimental MS/MS spectral data is the database search. In this technique, the experimental MS/MS spectral data are searched (compared) against a database of simulated (also called model or theoretical) spectra aiming to find the true- or the closest- match available in the database. The simulated spectra database is constructed by in-silico simulation of shotgun proteomics experiment using a proteome sequence database and known post-translational modifications (PTMs).
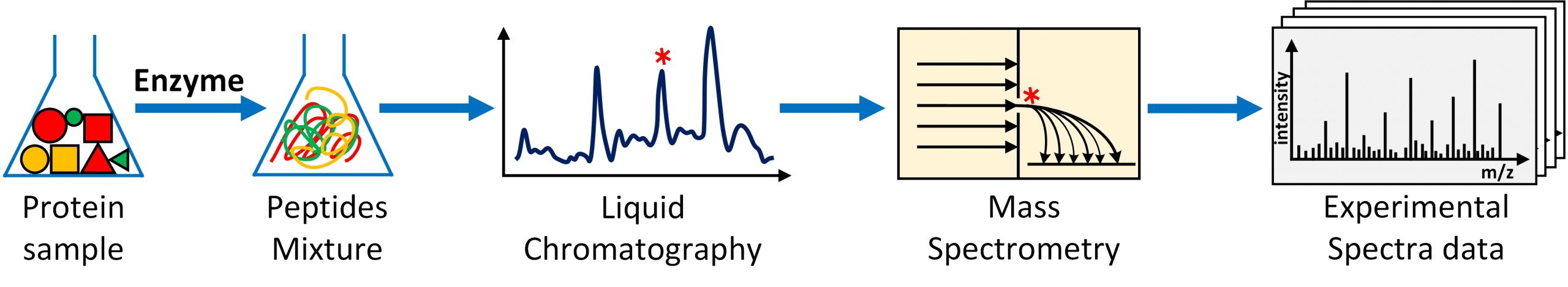
Figure 1. Generating experimental MS/MS spectral data from a protein sample through shotgun proteomics experiment.
The above explained problem can also be understood as a ‘face-in-a-crowd’ or ‘needle-in-a-haystack’ problem. Consider that each of the experimental MS/MS spectrum is the photo of (supposedly) a human face. Then, the constructed database will contain the images of all available (human) faces and the problem would be to find the exact or the best possible match for each photo in the experimental dataset. Let us say that about 1 million photos need to be identified against a database of photos of all ~7 billion people on Earth. Sounds fairly simple, right? Now let’s consider the case where the photos in the experimental dataset may have ‘mutations’. For example, photos of faces wearing hats, ear and/nose ornaments etc. and combinations of these. Then, to correctly identify these faces, the database must also be expanded to contain all or most possible mutations of the 7 billion entries present in the database inflating the database size to several hundred billion photos. Same is the case with database peptide search that to correctly identify the experimental MS/MS spectra containing mutations (for example, peptides in a tumor or cancer sample), the simulated spectra database must also be appended with the mutated versions of the simulated spectra inflating its size to several hundred GBs to a few TBs.


In the past, the prime focus of research in database search has been to speed up the search by reducing the number of computations required to identify the experimental MS/MS spectra. This effort included the invention of novel and highly impactful algorithms for database filtration, spectrum-to-spectrum comparisons and false discovery rate. For example, considering the ‘face-in-a-crowd’ analogy, given a photo of a face that looks like an Asian female individual, the database of interest may be narrowed down to only the photos of Asian female population and so on. However, owing to the size and nature of experimental MS/MS data and the simulated database, the performance of the existing state-of-the-art algorithms is upper-bounded by:
- The overheads associated with communicating the data from its memory source to the target processing element within the hierarchy.
- The amount of compute and memory resources required.
Consequently, these algorithms may require several hours to months of computational work to complete a database peptide search experiment depending on the size of problem (i.e. experimental dataset and the simulated spectra database).
Proposed Research
Our current research is focused towards the design and development of a distributed-memory parallel computational software, called HiCOPS, that is capable of efficiently (and scalably) accelerating the state-of-the-art database peptide search algorithms on modern HPC supercomputing architectures. The HiCOPS design is based on the SPMD Bulk Synchronous Parallel computational model. The proposed HiCOPS software workflow can be oversimplified into four main supersteps (See Figure 2):
- Split the algorithmic workload among the supercomputer’s processing nodes in a load balanced fashion
- Pre-process, convert, index and load the experimental MS/MS dataset in a data parallel fashion.
- Independently executes the partial database peptide search on each computing node for the complete experimental MS/MS dataset.
- Merge the partial results at each computing node in an communication optimal way.
This allows the HiCOPS to alleviate all resource and overhead (communication) upper bounds that exist in the current generation of (shared-memory based) database peptide search methods and software. Several optimization techniques including buffering, sampling, overlapped computations and communications and task scheduling have been implemented to aid in optimal parallel performance. Further, the HiCOPS’ split workload design allows for a much improved performance-per-node yield with increasing number of nodes due to the reduced memory-contention (communication) cost associated with database and data movement and management within each node yielding hyper-linear speedups.
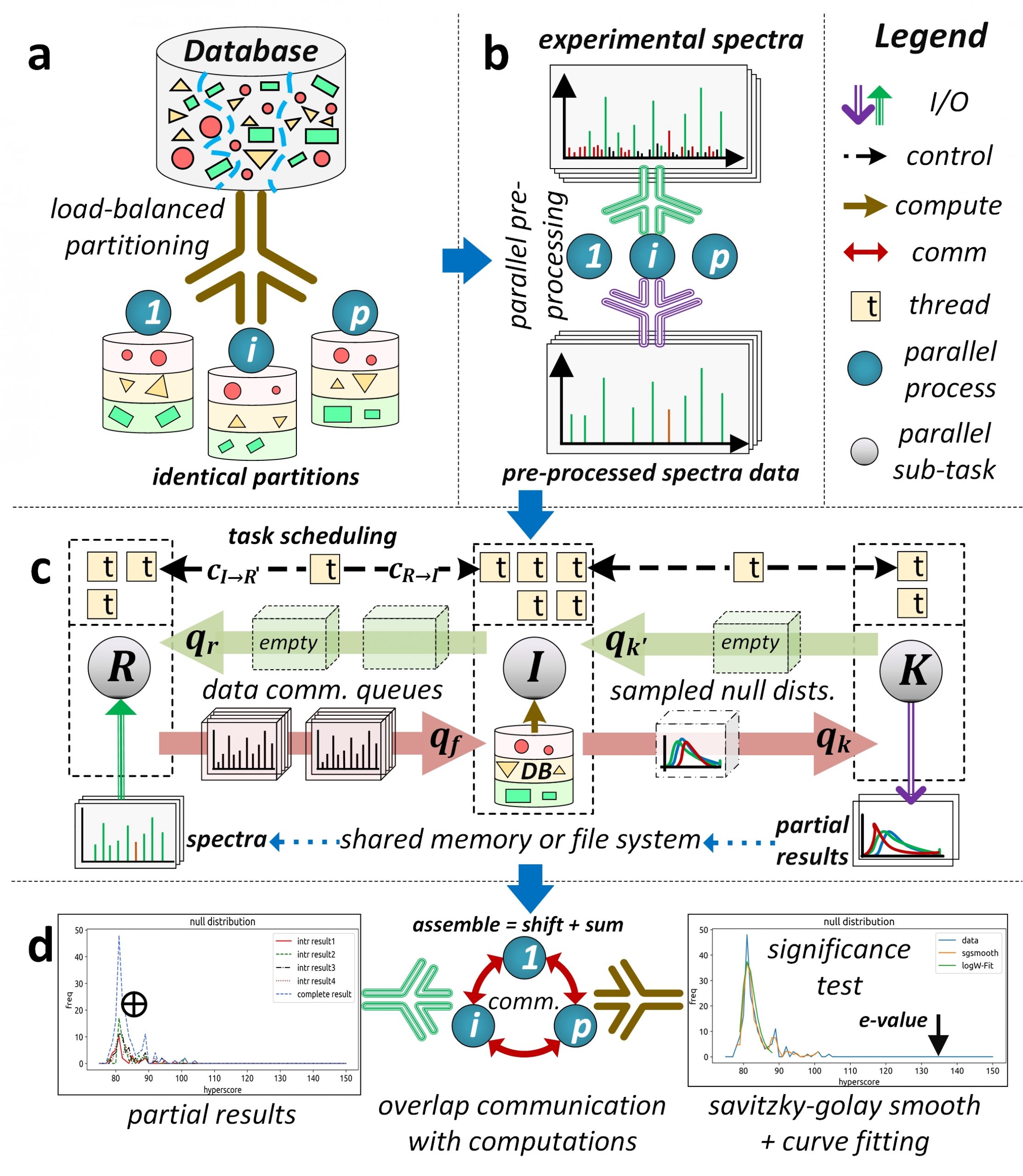
Figure 2. Oversimplified depiction of the split-compute-merge design strategy of HiCOPS parallel database peptide search software.
Results
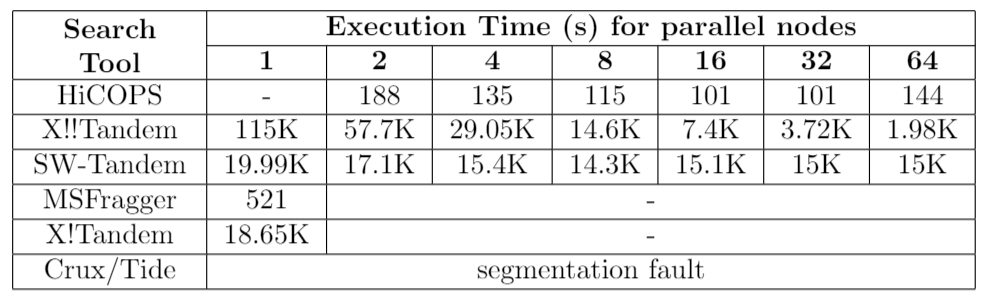
HiCOPS enables searching several GBs of experimental MS/MS spectra data to be searched against TBs of simulated spectra database(s) in few minutes; something not feasible even with weeks of computations with the existing state-of-the-art methods. (See Figure 3). We compared the HiCOPS with several existing serial (X!Tandem, Crux/Tide, MSFragger) and HPC (X!!Tandem, Bolt, SW-Tandem) software tools under varying experimental setting and found that the HiCOPS outperforms them by more than 100x in many cases and up to more than 500x as the experimental workloads increase as depicted in the following tables.
Table 1. Comparison between HiCOPS and existing tools for dataset size: 3.8 million, database size: 93.5 million. X!!Tandem and SW-Tandem ran for two days on Comet cluster (job limit) but did not complete their execution.
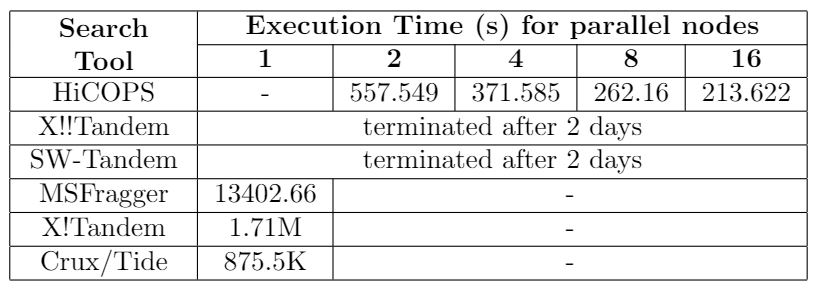
Table 2. Comparison between HiCOPS and MSFragger for extreme-scale experiments. The exp. 2 involved processing 1.5 million experimental spectra against a database of 1.6 billion peptides. MSFragger took 35.5 days to complete while HiCOPS completed in 103 minutes (72 nodes).
Performance Evaluation
The HiCOPS parallel performance and overhead costs were measured and analyzed using 12 experiment sets of varying workload sizes (varying database and dataset sizes). The analysis of results obtained for several key metrics including speedup scalability, hardware counters per node, overhead costs associated with load imbalance, communication, I/O and task-scheduling revealed near-optimal performance results and minimal overhead costs making HiCOPS an ideal foundation for the future generation of accelerated database peptide search. (See Figures 3-5 for results)
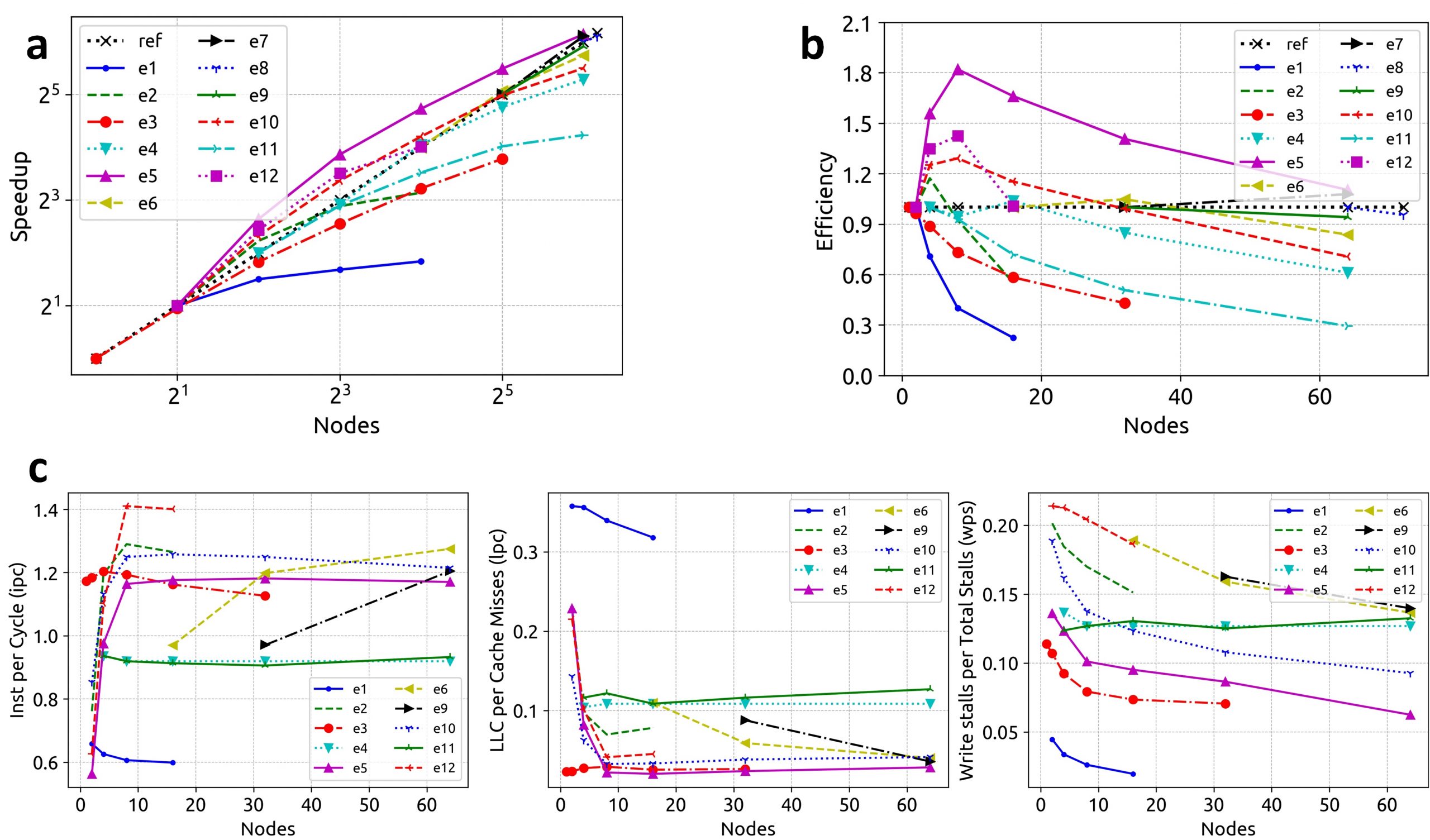
Figure 3. a, b) The performance improves as the workload size increases. c) Hardware performance (per node) for the 12 experiment sets. The instruction-per-cycle performance improves as the number of nodes are increased until saturated corresponding to hyper-linear speedups observed in Figures 3a and 3b.
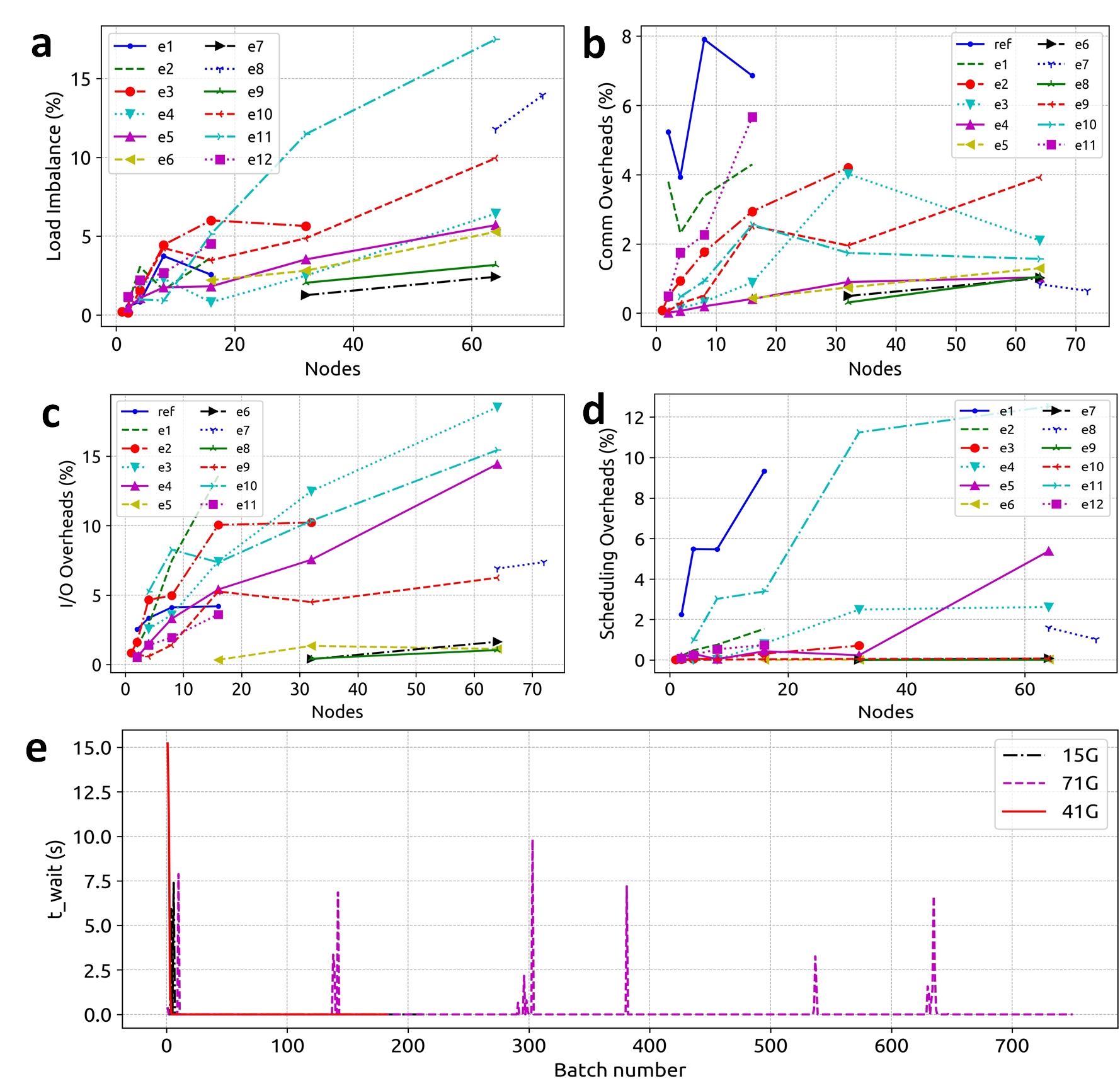
Figure 4. a) Percentage total runtime cost associated with load imbalance remains <10%. b) Percentage total runtime cost associated with inter-node communication remains <5%. c) Percentage total runtime cost associated with I/O remains <10%. d, e) Percentage total runtime cost associated with task scheduling remains <2%.
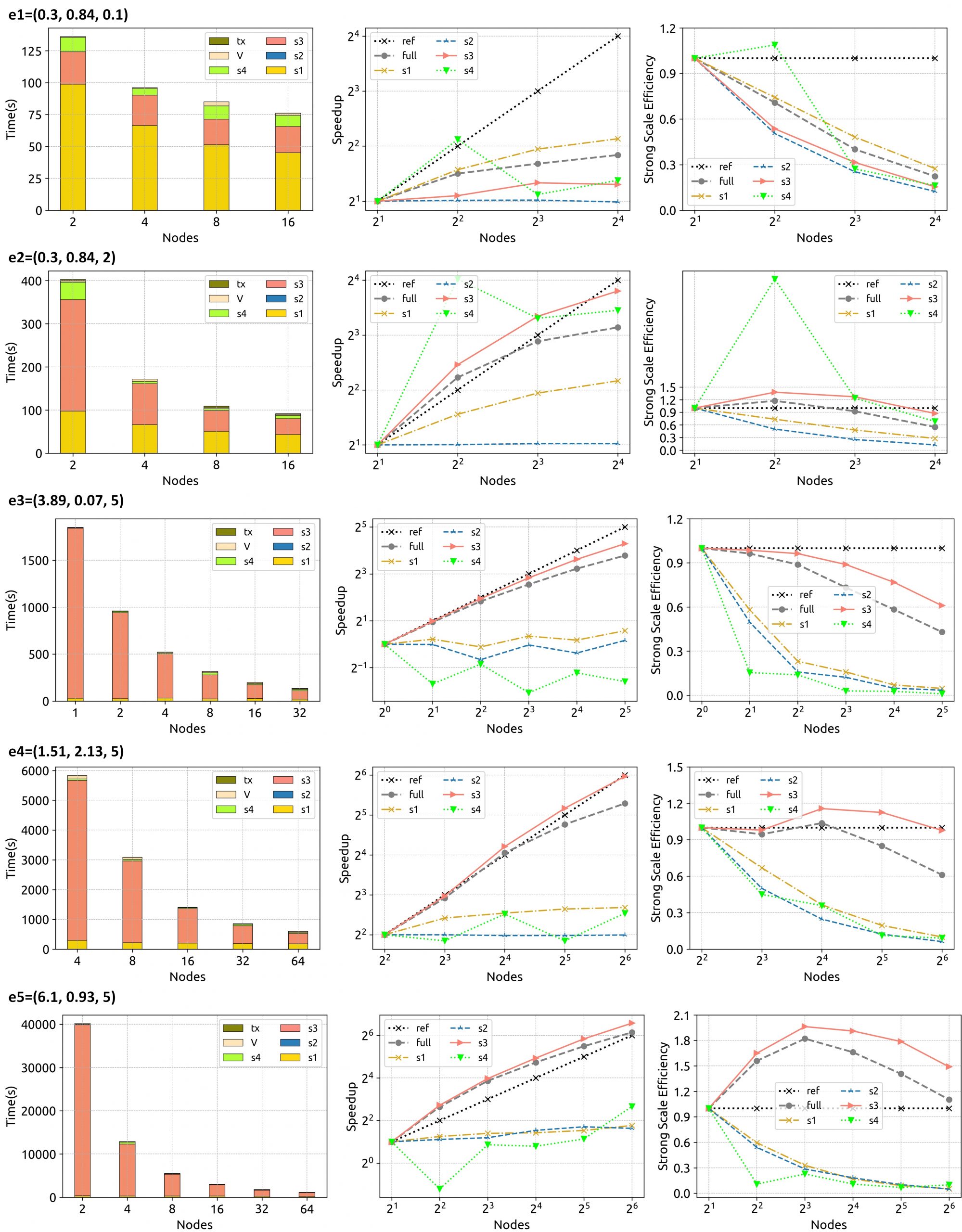
Figure 5. HiCOPS overall runtime, speedup scalability and efficiency (and its decomposition among supersteps and overheads) for each experiment set with increasing number of nodes depicts superior results for experiments with large workloads.
Conclusion
The existing research has primarily (successfully) focused towards improving the database peptide search by reducing the number of required computations which has led the overhead costs (communication) to upper-bound the performance that can be extracted from them. However, the recent trend in the database peptide search computational workloads associated with searching large volumes of publicly available MS/MS data against the humongous databases created with several post-translational modifications point towards an urgent need for software tools capable of harnessing hardware resources from supercomputing architectures. Our highly scalable and low-overhead parallel software, HiCOPS, meets this urgent need for the next generation of database peptide search application.
Recent Publications and Patents
1. Haseeb, Muhammad, and Fahad Saeed. “High performance computing framework for tera-scale database search of mass spectrometry data.” Nature Computational Science 1, no. 8 (2021): 550-561 Nature
2. Saeed, Fahad, and Muhammad Haseeb*. “Methods and systems for compressing data.” U.S. Patent 10,810,180, issued October 20, 2020. Google Patents
3. Haseeb, Muhammad*, and Fahad Saeed. “Efficient Shared Peak Counting in Database Peptide Search Using Compact Data Structure for Fragment-Ion Index.” In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp. 275-278. IEEE, 2019. Xplore
4. Haseeb, Muhammad*, Fatima Afzali, and Fahad Saeed. “LBE: A Computational Load Balancing Algorithm for Speeding up Parallel Peptide Search in Mass-Spectrometry based Proteomics.” In 2019 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW), pp. 191-198. IEEE, 2019. Xplore