Historically, there have been two contrasting approaches for inferring peptides from mass-spectrometry data, i.e., de novo sequencing and database searching. The de novo approach tries to transform spectral space into peptide space by predicting individual amino acids from a given spectrum. On the other hand, database search tries to associate the experimental spectra to existing peptides by transforming peptide space into the spectral space and performing the comparisons. Each approach uses a heuristic similarity-scoring function to determine the match quality between an experimental spectrum and its corresponding peptide. However, when using heuristics, there is no solid reasoning outlining why a function is chosen over the other or why a particular feature within a process has the given associated weight. On the other hand, theoretical spectra are usually generated using a simple form of a simulator, introducing another source of inaccuracy in the search process.
Here we will discuss the design and implementation of a deep learning model called SpeCollate, which overcomes these issues by directly learning the similarity function between experimental spectra and peptide strings. SpeCollate transforms spectral and peptidal spaces into a shared Euclidean subspace by learning embeddings for spectra and peptides. The L2 distance between two data points in the resultant space directly correlates with their similarity.
By training the network on nearly 4.8 million sextuplets, obtained from the NIST and MassIVE peptide libraries, SpeCollate can achieve a promising peptide identification accuracy of up to ~99%.
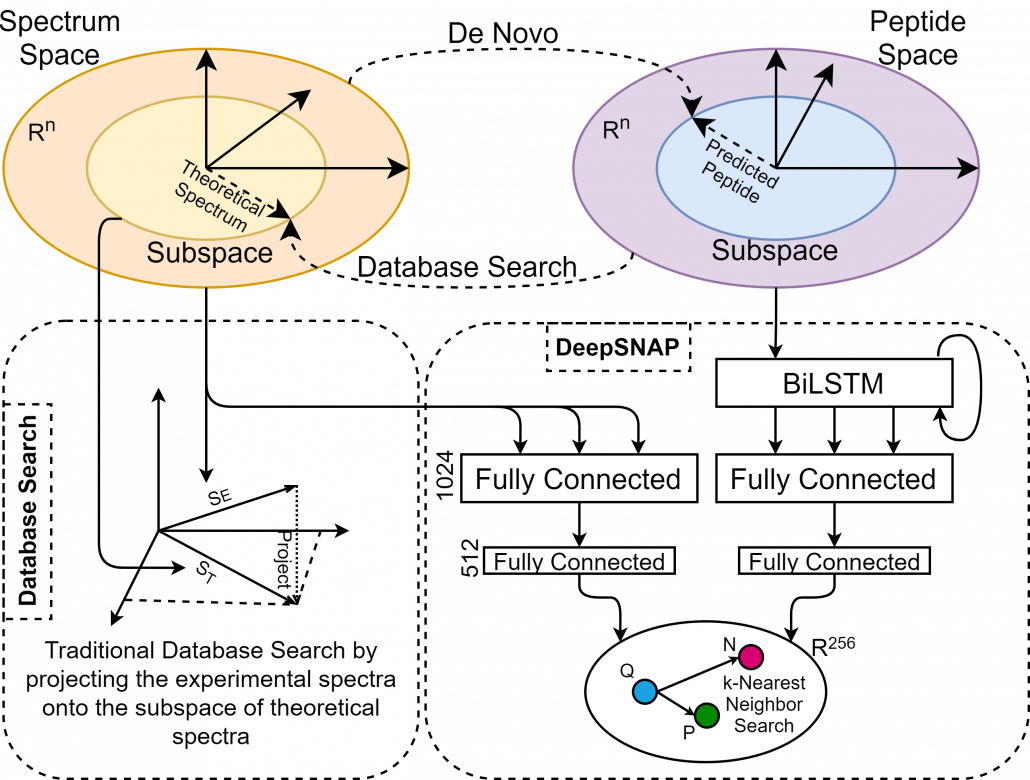
Fig 1. De novo and database search, which try to transform one space into another. This is prone to error and uncertainty as a lot of information can be missed. On the contrary, SpeCollate learns the same sized embeddings for both peptides and spectra by projecting them to shared Euclidean space.
Deep Learning methods are the next step in Big Data Proteomics. The limitations and oversights of the existing numerical techniques, bounded performance of spectral simulators, unoptimized scoring heuristics, inflated search space, and the opportunities made available by huge data repositories with annotated spectra are some of the key motivators behind this paradigm shift. Currently, there is no single heuristic from database search techniques that can claim as the most accurate strategy. Substantial work has been carried out towards developing computational methods for identifying peptides using database search and de novo algorithms. However, peptide identification problems are well-known and prevalent, including but not limited to misidentifications or no identifications for peptides, statistical accuracy (FDR), and inconsistencies between different search engines. Comparison across literature indicates decreased average accuracy of de novo algorithms (<35%) relative to database search algorithms (30-80%). Lack of quality assessment benchmarks makes the accuracy exhibited from these database search tools highly dependent on the data, indicating that further formal investigation and evaluation is warranted. Two significant sources of heuristic errors introduced in the numerical database search algorithms are how the peptide deduction takes place, i.e., simulation of the spectra (from peptides) and the peptide spectrum match scoring-function. The simplistic and a priori nature of the scoring mechanism neglects the MS data (and the database) that are under consideration, leading to variable quality peptide deductions.
We make two fundamental contributions in MS-based proteomics search:
1) We present the design and implementation of Deep Similarity Network, called SpeCollate, for Proteomics Database Search. The goal is to learn a fixed-sized embedding of variable length experimental spectra and peptide strings so that the spectrum and its corresponding peptide are projected close to each other in a shared subspace. Our proposed network consists of two sub-networks, i.e., spectrum sub-network (SSN), which comprises two fully connected layers, and peptide sub-network (PSN), which consists of one bi-directional LSTM followed by two fully connected layers.
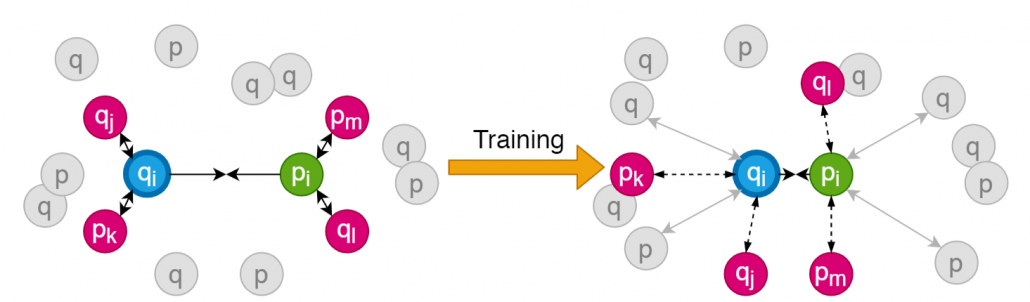
Fig 2. Training process using SNAP-loss function. The anchor and the positive peptide are moved closer to each other at each iteration, while the negative spectra and peptides are moved farther from the positive pair. The positives and negatives are selected through online-mining for calculating loss.
2) We formulate and implement a custom loss function called SNAP-loss for training the proposed deep-similarity network SpeCollate. This training process takes sextuplets as input, consisting of a combination of positive, negative, and anchor spectra and peptides. Generalization of the network is accomplished by training the network through multitask-learning, where the network is trained to predict multiple relevant features, including the fragment ion sequences, precursor mass, charge state, and fragmentation processes. We train or network for 200 epochs on a dataset of size ~4.8 million sextuplets.
We measure the performance of our modal by ROC and Precision-Recall curves. When compared against traditional scoring functions, i.e., XCorr and Hyperscore, SpeCollate outperforms both with a significant margin.
Recent Publications:
1. Leyva, Dennys, Muhammad Usman Tariq, Rudolf Jaffé, Fahad Saeed*, and Francisco Fernandez Lima. “Unsupervised Structural Classification of Dissolved Organic Matter Based on Fragmentation Pathways.” Environmental science & technology (2022). ACS
2. Fahad Saeed*, and Muhammad Usman Tariq. “Systems and methods for measuring similarity between mass spectra and peptides.” U.S. Patent 11,251,031, issued February 15, 2022. US Patent
3. Usman Tariq, and Fahad Saeed* , “SpeCollate: Deep cross-modal similarity network for mass spectrometry data based peptide deductions“, PLoS ONE, Vol. 16, Issue 10, Oct 2021 PLoS
4. Muhammad Usman Tariq, Dennys Leyva, Francisco Fernandez Lima, and Fahad Saeed*, “Graph Theoretic Approach for the Analysis of Comprehensive Mass-Spectrometry (MS/MS) Data of Dissolved Organic Matter“, International Workshop on Biological Network Analysis and Integrative Graph-Based Approaches (IWBNA), Proceedings of IEEE International Conference on Bioinformatics and Biomedicine (BIBM), December 9-12, 2021